

Multi-Valent siRNAs for CNS Gene Silencing: Advancing Huntington’s Disease Therapy
Huntington’s disease (HD) is a rare, inherited neurodegenerative condition caused by an expanded CAG repeat in exon 1 of the HTT gene. Although individuals may carry the mutant allele for decades without symptoms, progression is inevitable.
For over a decade, we have worked with the CHDI Foundation and Dr. Neil Aronin’s lab to develop therapies aimed at delaying or even preventing disease onset. Our goal is a convenient therapeutic that preserves cognitive function and quality of life.
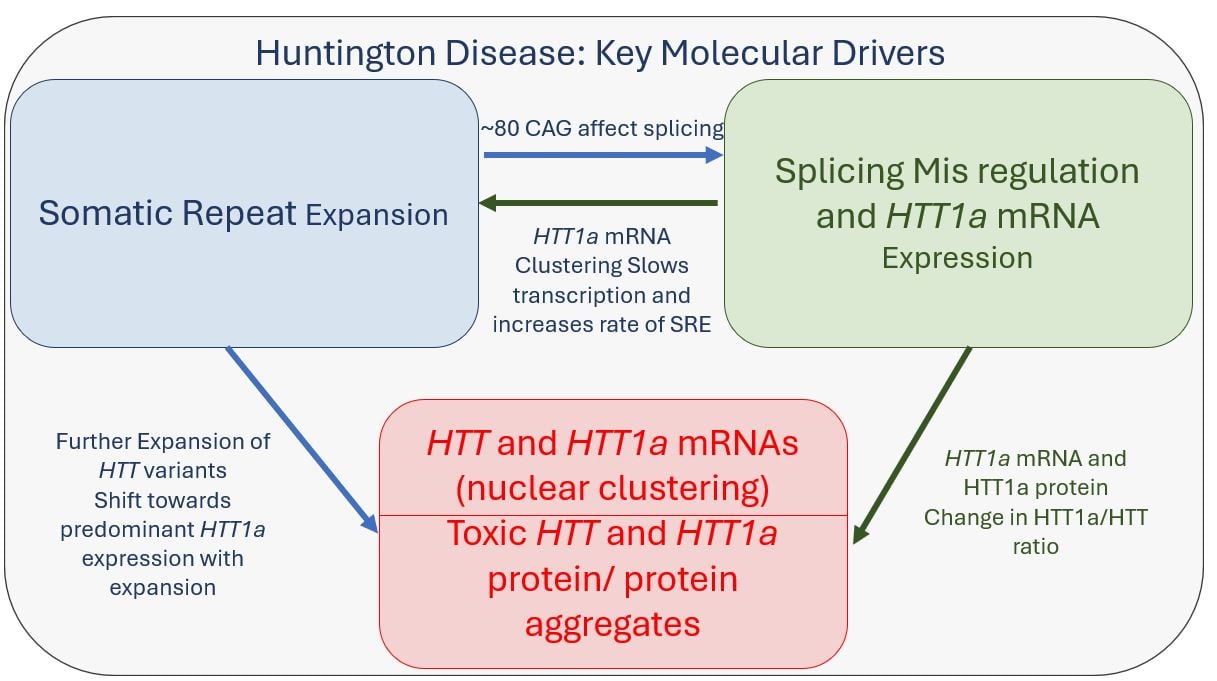
Recent advances from multiple laboratories have significantly deepened our understanding of HD’s molecular mechanisms. Three key pathological interconnected modules have emerged.
- Somatic repeat expansion, driven by mismatch repair pathway
- Splicing misregulation leading to the HTT1a isoform production
- Neurotoxicity from expanded polyglutamine tracts in both HTT1a and full-length HTT (proteins (particularly with >150 CAG repeats).
We have developed potent siRNAs that target both the full-length HTT transcript and MSH3, a key factor in repeat expansion. CNS-optimized di-siRNAs targeting MSH3 halted expansion and reversed neurodegenerative biomarkers in mouse models—even in the presence of highly expanded repeats.
Targeting HTT1a is more difficult due to its nuclear clustering near the transcription site, limiting accessibility to RNA-based drugs. We are collaborating with leading experts, such as the Gillian Bates Lab at University College London, to understand HTT1a biology and develop innovative targeting strategies. We are designing oligonucleotides that favor nuclear localization and are actively exploring combinatorial treatments to address both somatic instability and pathogenic transcript expression.