Feb 14, 2023
Coming up with new ways to treat COVID-19 is at the *heart* of BMB.
Read more


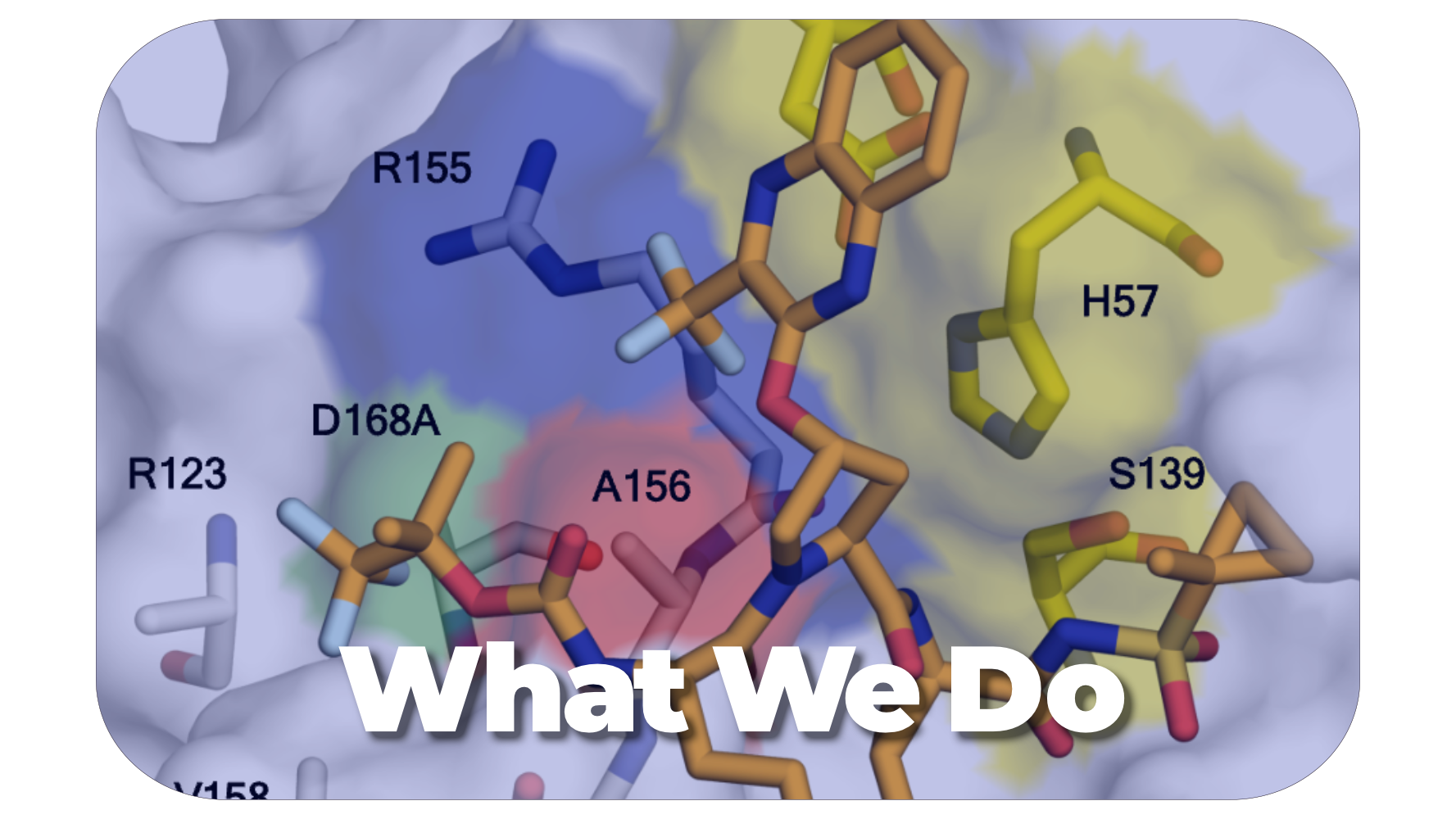
We develop small molecule antiviral lead compounds against HIV, HCV, DENV, SARS-CoV-2, and other pathogenic viruses.

Drug resistance is a major limitation in the treatment of infections and cancers, so understanding the molecular mechanisms of drug resistance could lead to more effective therapies.
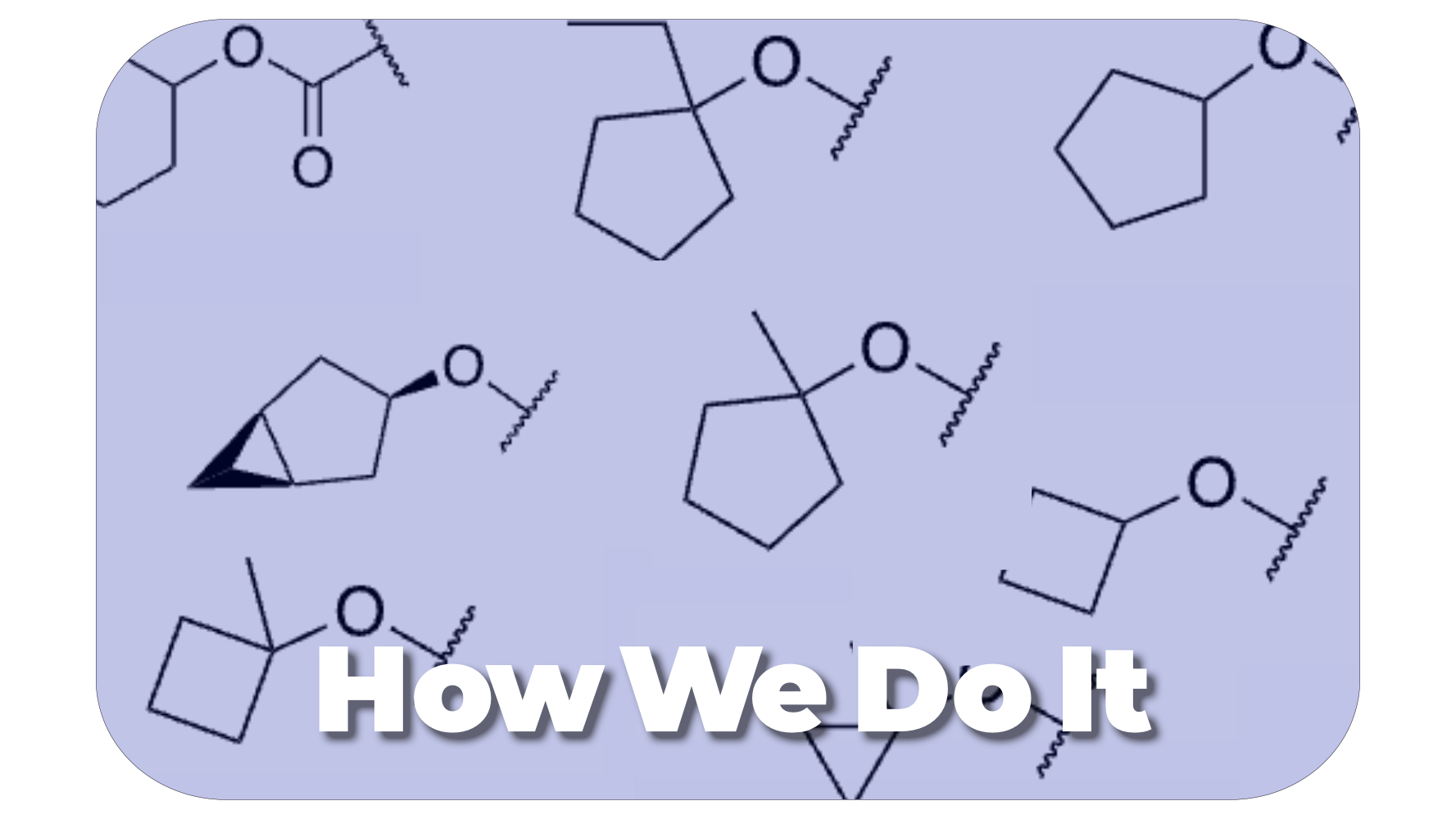
Our laboratory uses structure-based drug design and insights from our understanding of the mechanisms of drug resistance to rationally design antiviral compounds.

We are a team of organic/medicinal chemists with a passion for small molecule drug discovery.
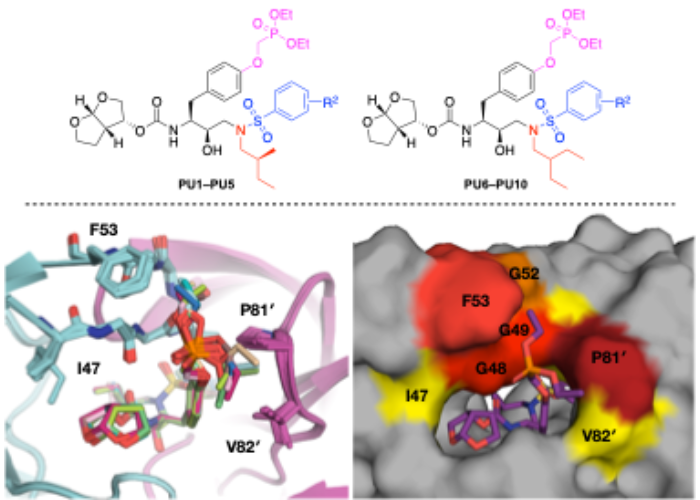 HIV-1 Protease Inhibitors with a P1 Phosphonate Modification Maintain Potency against Drug-Resistant Variants by Increased Interactions with Flap Residues.
HIV-1 Protease Inhibitors with a P1 Phosphonate Modification Maintain Potency against Drug-Resistant Variants by Increased Interactions with Flap Residues.
Lockbaum, G. J.; Rusere, L. N.; Henes, M.; Kosovrasti, K.; NageshwaraRao, D.; Lee, S-K.; Spielvogel, E.; Nalivaika, E. A.; Swanstrom, R.; Yilmaz, N. K.; Schiffer, C. A.; Ali, A. Eur. J. Med. Chem. 2023, 257, 115501 – https://doi.org/10.1016/j.ejmech.2023.115501
Protease inhibitors are the most potent antivirals against HIV-1, but they still lose efficacy against resistant variants. Improving the resistance profile is key to developing more robust inhibitors, which may be promising candidates for simplified next-generation antiretroviral therapies. In this study, we explored analogs of darunavir with a P1 phosphonate modification in combination with increasing size of the P1′ hydrophobic group and various P2′ moieties to improve potency against resistant variants. The phosphonate moiety substantially improved potency against highly mutated and resistant HIV-1 protease variants, but only when combined with more hydrophobic moieties at the P1′ and P2′ positions. Phosphonate analogs with a larger hydrophobic P1′ moiety maintained excellent antiviral potency against a panel of highly resistant HIV-1 variants, with significantly improved resistance profiles. The cocrystal structures indicate that the phosphonate moiety makes extensive hydrophobic interactions with the protease, especially with the flap residues. Many residues involved in these protease-inhibitor interactions are conserved, enabling the inhibitors to maintain potency against highly resistant variants. These results highlight the need to balance inhibitor physicochemical properties by simultaneous modification of chemical groups to further improve resistance profiles.
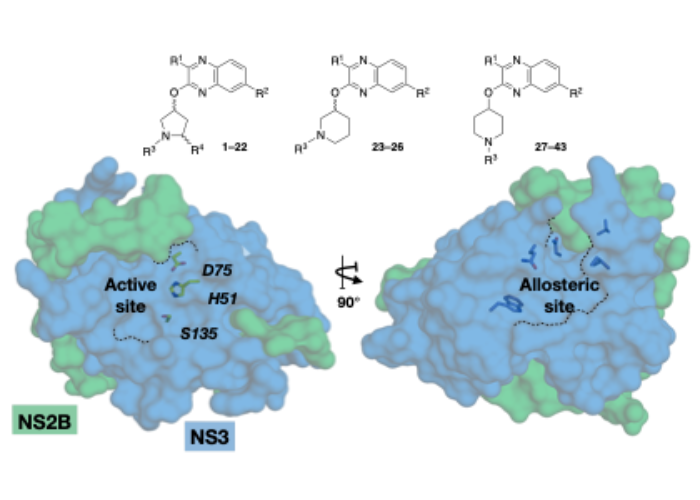 Allosteric Quinoxaline-Based Inhibitors of the Flavivirus NS2B/NS3 Protease.
Allosteric Quinoxaline-Based Inhibitors of the Flavivirus NS2B/NS3 Protease.
Zephyr, J.; Nageswara Rao, D.; Johnson, C.; Shaqra, A. M.; Nalivaika, E.; Jordan, A.; Kurt Yilmaz, N.; Ali, A.; Schiffer, C. A.Bioorg. Chem. 2023, 131, 106269 – https://doi.org/10.1016/j.bioorg.2022.106269
Viruses from the Flavivirus genus infect millions of people worldwide and cause severe diseases, including recent epidemics of dengue virus (DENV), and Zika virus (ZIKV). There is currently no antiviral treatment against flavivirus infections, despite considerable efforts to develop inhibitors against essential viral enzymes including NS2B/NS3 protease. Targeting the flavivirus NS2B/NS3 protease proved to be challenging because of the conformational dynamics, topology, and electrostatic properties of the active site. Here, we report the identification of quinoxaline-based allosteric inhibitors by fragment-based drug discovery approach as a promising new drug-like scaffold to target the NS2B/NS3 protease. Enzymatic assays and mutational analysis of the allosteric site in ZIKV NS2B/NS3 protease support noncompetitive inhibition mechanism as well as engineered DENV protease construct indicating the compounds likely compete with the NS2B cofactor for binding to the protease domain. Furthermore, antiviral activity confirmed the therapeutic potential of this new inhibitor scaffold.
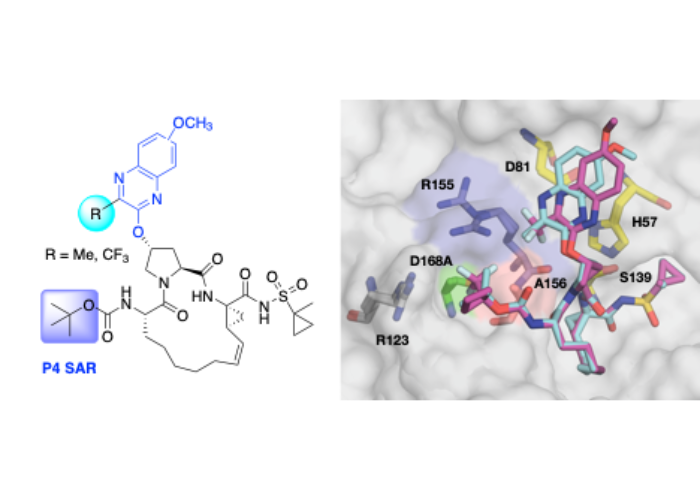 Discovery of Quinoxaline-Based P1–P3 Macrocyclic NS3/4A Protease Inhibitors with Potent Activity against Drug-Resistant HCV Variants.
Discovery of Quinoxaline-Based P1–P3 Macrocyclic NS3/4A Protease Inhibitors with Potent Activity against Drug-Resistant HCV Variants.
Nageswara Rao, D.; Zephyr, J.; Henes, M.; Chan, E. T.; Matthew, A. N.; Hedger, A. K.; Conway, H. L.; Saeed, M.; Newton, A.; Petropoulos, C. J.; Huang, W.; Yilmaz, N. K.; Schiffer, C. A.; Ali, A.Med. Chem. 2021, 64, 11972–11989. https://pubs.acs.org/doi/full/10.1021/acs.jmedchem.1c00554
The three pan-genotypic HCV NS3/4A protease inhibitors (PIs) currently in clinical use—grazoprevir, glecaprevir and voxilaprevir—are quinoxaline-based P2–P4 macrocycles and thus exhibit similar drug resistance profiles. Using our quinoxaline-based P1–P3 macrocyclic lead compounds as an alternative chemical scaffold, we explored structure-activity relationships (SARs) at the P2 and P4 positions to develop pan-genotypic PIs that avoid drug resistance. A structure-guided strategy was used to design and synthesize two series of compounds with different P2 quinoxalines in combination with diverse P4 groups of varying size and shape, with and without fluorine substitutions. Our SAR data and cocrystal structures revealed an interplay between the P2 and P4 groups which influenced inhibitor binding and overall resistance profile. Optimizing inhibitor interactions in the S4 pocket led to PIs with excellent antiviral activity against clinically relevant PI-resistant HCV variants and genotype 3, providing potential pan-genotypic inhibitors with improved resistance profile.