


HCV Protease
The generality of the substrate envelope hypothesis have been evaluated by investigating other viral systems such as HCV NS3/4A serine protease. Crystal structures of protease-substrate complexes reveal that viral substrates bind to the protease active site in a conserved manner defining the NS3/4A substrate envelope. (Romano et. al., 2010)
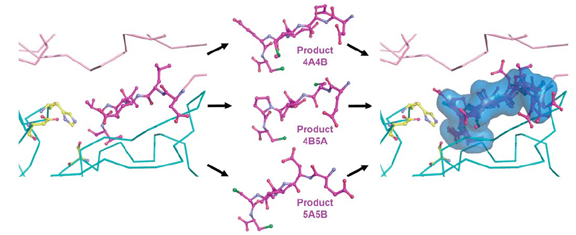
Product complexes 4A/4B, 4B/5A and 5A/5B were superimposed onto the full-length NS3/4A structure (1CU1) to reveal the conserved mode of binding.
Drug resistance in HCV NS3/4A Protease
Resistance against the most promising NS3/4A protease inhibitors has emerged in clinical trials. The majority of reported drug resistance mutations cluster around the protease active site.
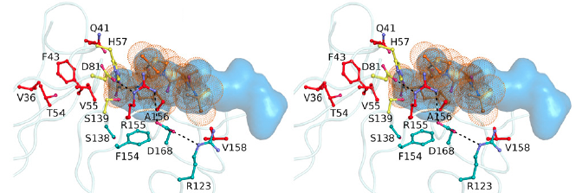
Mutations that confer the most severe resistance in the clinic occur at sites where the inhibitors protrude from the substrate envelope.
Crystal structures of inhibitor bound NS3/4A protease validate the substrate envelope hypothesis as a general model for predicting the susceptibility of protease inhibitors to resistance. Inhibitors designed to fit within the substrate envelope will be less susceptible to resistance, as mutations affecting inhibitor binding would simultaneously interfere with the recognition of viral substrates.
Targeting HCV NS3/4A Protease
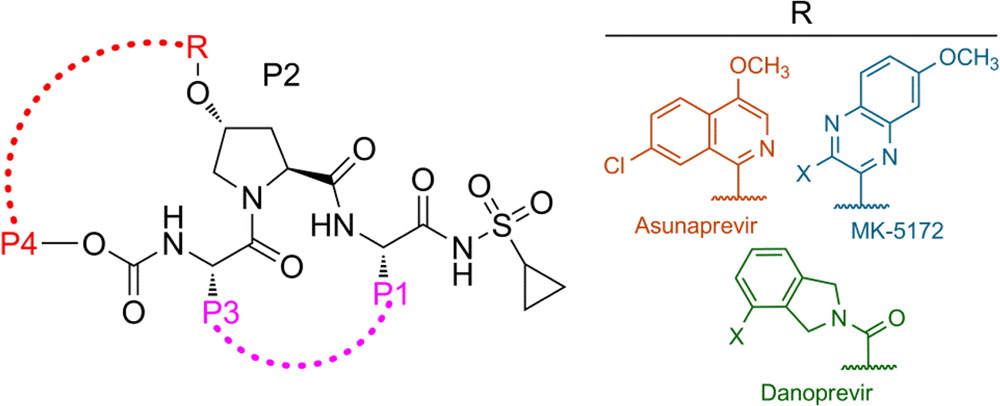
The available antiviral therapies, which include PEGylated interferon, ribavirin, and one of the HCV NS3/4A protease inhibitors telaprevir or boceprevir, are ineffective for some patients and cause severe side effects. More potent NS3/4A protease inhibitors are in clinical development, but the long-term effectiveness of these drugs is challenged by the development of drug resistance. Recently, we investigated the role of macrocycles in the susceptibility of NS3/4A protease inhibitors to drug resistance in asunaprevir, danoprevir, vaniprevir, and MK-5172, with similar core structures but varied P2 moieties and macrocyclizations. Linear and macrocyclic analogues of these drugs were designed, synthesized, and tested against wild-type and drug-resistant variants R155K, V36M/R155K, A156T, and D168A in enzymatic and antiviral assays. Macrocyclic inhibitors were generally more potent, but the location of the macrocycle was critical for retaining activity against drug-resistant variants: the P1–P3 macrocyclic inhibitors were less susceptible to drug resistance than the linear and P2–P4 macrocyclic analogues. In addition, the heterocyclic moiety at P2 largely determined the inhibitor resistance profile, susceptibility to drug resistance, and the extent of modulation by the helicase domain. Our findings suggest that to design robust inhibitors that retain potency to drug-resistant NS3/4A protease variants, inhibitors should combine P1–P3 macrocycles with flexible P2 moieties that optimally contact with the invariable catalytic triad of this enzyme (Ali et al., 2013).
 |
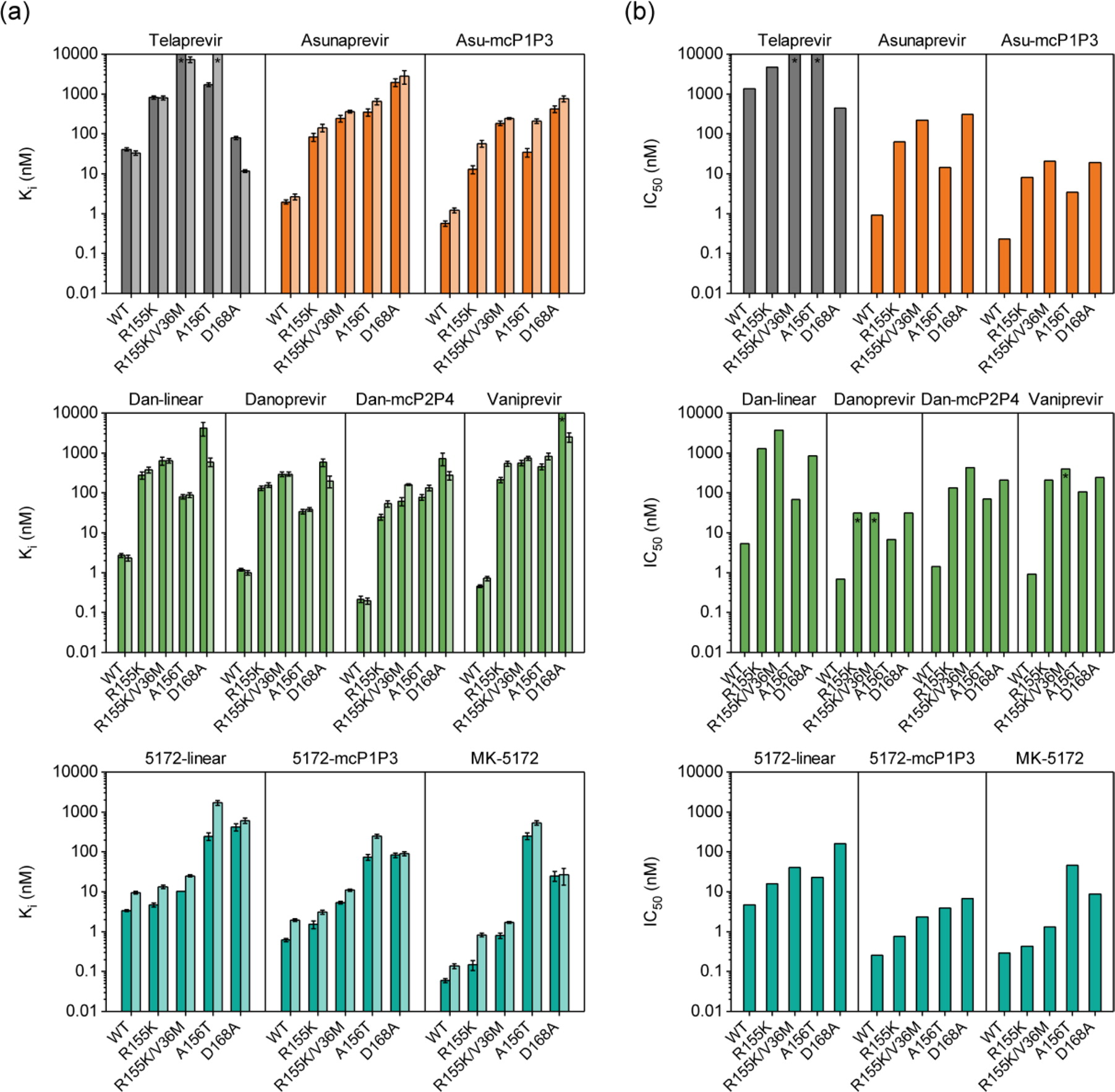 |
| Enzyme inhibition constants for HCV genotype 1a full-length NS3/4A and isolated protease domain (dark and light bars) and (b) replicon-based half maximal inhibitory concentrations for genotype 1b HCV NS3/4A and drug-resistant variants for (first row) telaprevir and asunaprevir series, (second row) danoprevir/vaniprevir series, and (third row) MK-5172 series. Error bars represent standard errors of the mean (n = 4); * indicates Ki and IC50 values are greater than the highest inhibitor concentration tested (Ali et al., 2013). |
|